Background: Current B cell-directed MS therapies affect a heterogeneous B cell compartment in a global manner. However, functionally distinct B cell subsets impact differently on MS pathogenesis. The specification pathways of MS-relevant B cell pools and their phenotypic characterization are unclear. This knowledge gap prevents personalized MS treatments.
Hypothesis: B-lymphoid subsets with pro- or anti-inflammatory phenotypes can be distinguished and monitored by individual molecular marker profiles and transcriptional signatures, and their dynamic (re-) appearance can be associated with checkpoints of MS pathology in the absence or presence of treatments.
Strategy: We will establish a B cell inventory of MS patients and EAE mouse models under various treatment conditions by applying a combination of cellular and molecular phenotyping with a special focus of genetic B cell programming through the Transcription Factor EB (TFEB), which we recently identified as signal checkpoint defining B cell fate decisions.
Summary
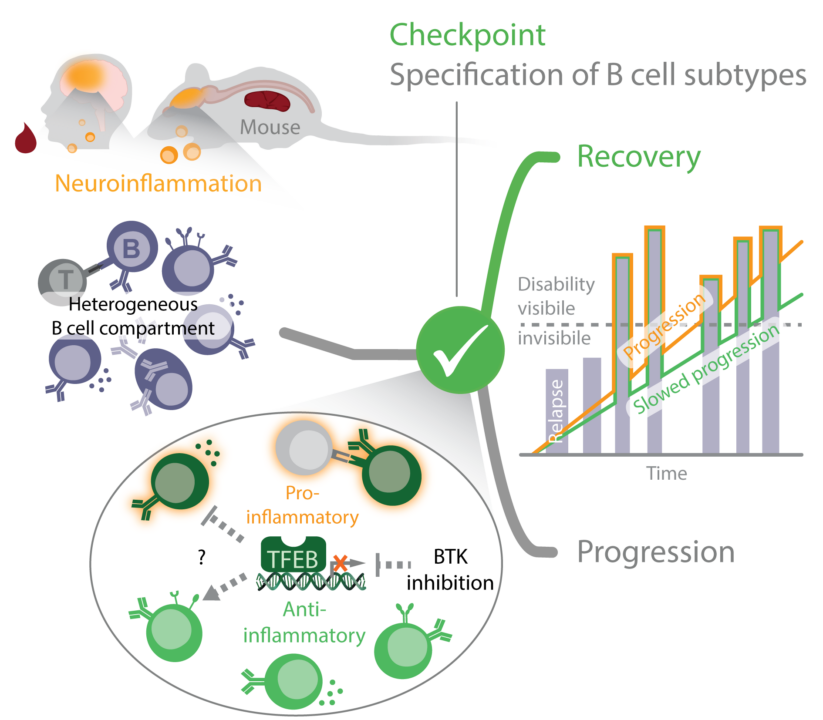
For decades, multiple sclerosis (MS) has been thought to represent a mainly T cell-mediated autoimmune disease. However, compelling evidence obtained from clinical studies as well as experimental animal models more recently unveiled key contributions of B lymphocytes to MS pathogenesis. Not only the production of antibodies but also (auto) antigen presentation and B-lymphoid secretion of pro-inflammatory cytokines drive MS progression and thus, hinder CNS recovery from acute inflammatory bouts. Inversely, therapeutic B cell depletion by clinical administration of anti-CD20 antibodies, which eliminate peripheral B cell subpopulations, dampens the inflammation of the central nervous system (CNS) and can prevent MS relapses. However, B cells or B cell subpopulations may also ameliorate chronic CNS inflammation through the production of anti-inflammatory cytokines, most notably IL-10. It thus appears that individual B cell subpopulations with functionally opposing properties exist and that accordingly, B cells may serve as a cellular checkpoint for CNS recovery in MS. However, a phenotypic characterization of MS-relevant B cell pools is still out, but is urgently needed to optimize MS treatment options. Our project proposal aims at a comprehensive inventory of B cell compartments with pro- or anti-inflammatory properties in acute relapse biology and progression of MS. We will also study B-lymphoid phenotypes in blood samples of MS patients before and after the replenishment of the B cell repertoire throughout the course of anti-CD20 treatment. This part will shed light on the controversially discussed contribution of newly generated and/or antigen-experienced memory B cells to the MS pathology. A special focus will be given to the activation of the Transcription Factor E Box-binding (TFEB) that we discovered in our preparatory studies as a novel signal checkpoint of B cell activation and a phenotypic marker for certain memory B cell compartments in humans. Our recently generated mouse mutant with B cell-specific ablation of TFEB allows us to test for transcriptional signatures defining the functional specification of B cells in autoimmune processes. These investigations will be conducted in the presence or absence of B cell-modifying pharmaceuticals such as inhibitors of Bruton’s Tyrosine Kinase (BTK), which suppress B cell receptor signalling, including TFEB activation. In essence, the proposed study will index individual B cell populations of the naïve and memory compartments during different phases of MS and assess how the B cell compartment can be therapeutically shaped in a desirable manner.




