Background: Neuronal function and resilience in the inflamed CNS are impaired by an “energy crisis” that is due to mitochondrial dysfunction and ionic overload.
Hypothesis: We hypothesize that specific metabolic pathways might be neuronal cell type- and compartment-specific checkpoints that contribute to the “energy crisis” and govern the balance between damage and recovery in the inflamed white and grey matter.
Strategy: Using in vivo biosensor imaging, translatomics and proteomics profiling, metabolic modeling and molecular interventions, we will identify and attempt to rectify neuroenergetic deficits in multiple sclerosis and its animal models.
Summary
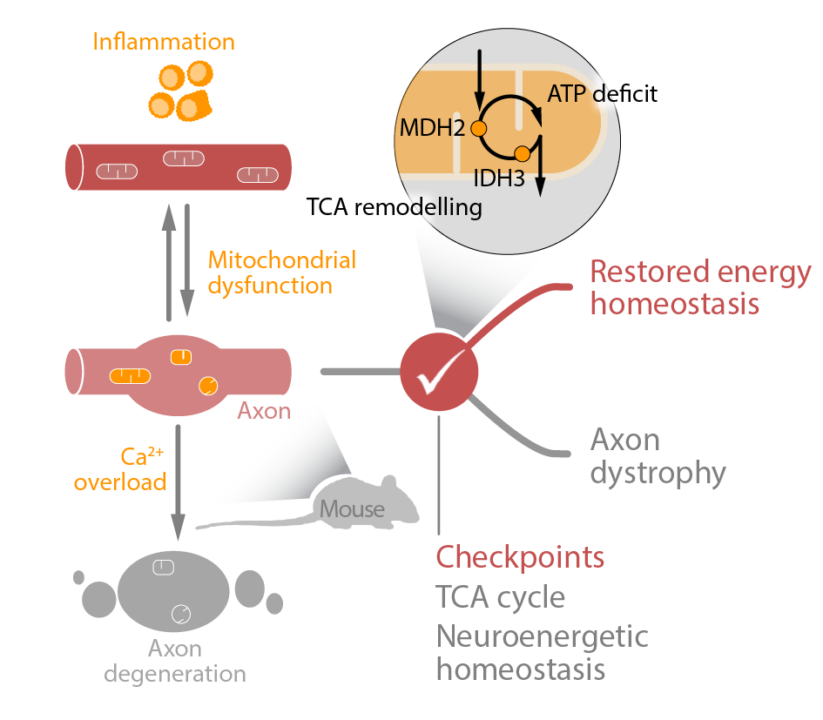
Multiple Sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) that results in neurodegeneration in the white, as well as in the grey matter. In this condition inflammation results in an impaired neuronal calcium homeostasis, which increases energy demand, while disturbances of mitochondrial function limit energy supply. The resulting neuronal “energy crisis” is widely considered to contribute to neuronal dysfunction and ensuing degeneration in both MS and its animal model. However, the molecular switches that govern neuronal energy balance in the inflamed CNS are currently unknown. In the first funding period, we have studied the mechanisms of neuronal resilience and identified a dysregulation of the tricarboxylic acid (TCA) cycle and more specifically the depletion of its pacemaker enzyme isocitrate dehydrogenase (IDH) 3 as a critical checkpoint of neuronal energy homeostasis both in experimental and human neuroinflammatory lesions. Notably, viral gene delivery of IDH3 was able to reverse the axonal energy deficits in a murine MS model. This reversal, however, was only partial, suggesting that existence of further neuroenergetic checkpoints that together determine the neuronal response to inflammation. Furthermore, it is currently unclear if similar or divergent checkpoints act in axons (primarily affected in white matter lesions) vs. other neuronal compartments such as soma or dendrites (a major target in the inflamed grey matter) and if such checkpoints are conserved across different neuronal cell populations. Finally, if we want to assess the therapeutic potential of such “neuroenergetic” manipulations, we need to resolve the reciprocal interactions between neuronal metabolism and neuronal function. In the upcoming funding period, we therefore want to identify and modulate neuroenergetic checkpoints in white matter (Aim ❶) and grey matter (Aim ❷) inflammation. For this, we will perform a comprehensive characterization of metabolic checkpoints in inflammatory lesions of the white and grey matter based on new tools for cell type-specific omics and bioenergetics characterization. We will combine use of genetically encoded metabolic biosensorsthat can reveal compartment-specific alterations with in vivo calcium imaging to record neuronal activity, as well as chemogenetic tools to modulate activity patterns. Finally, we want to use targeted molecular and metabolite manipulations to bolster neuronal energy homeostasis and assess the effects on neurodegenerative sequels and functional phenotypes. Continuing this project will enable us to better define the neuroenergetic checkpoints that govern neuronal resilience and recovery capacity in response to an inflammatory CNS attack.




