Summary
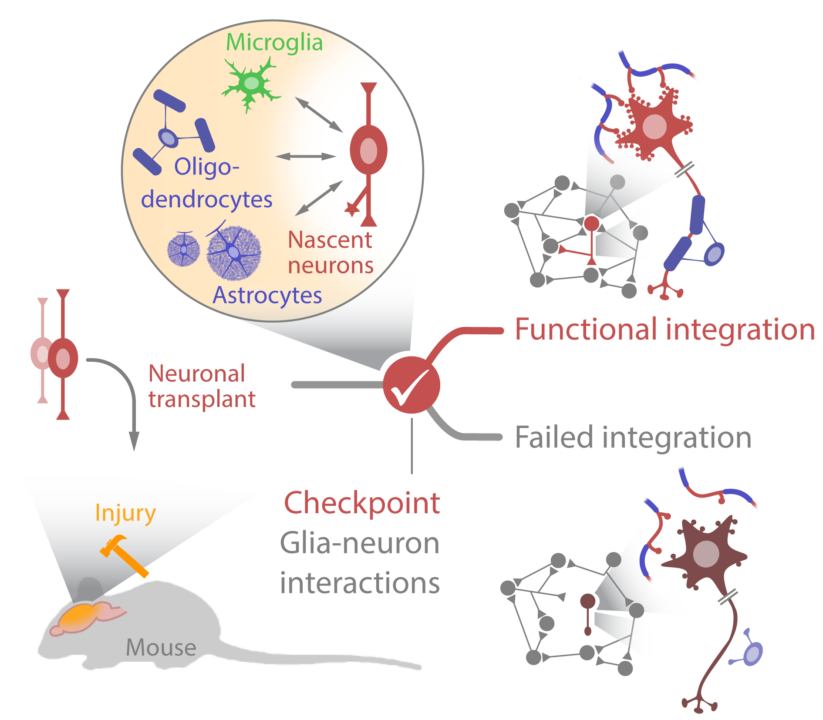
Neuronal loss in acute or chronic brain disease could be treated by neuronal replacement strategies, such as transplantation of young neurons, if they integrate properly into the existing networks. Here we explore the cell-based and molecular mechanisms causing hyperinnervation and overshoot loss of input connectivity as well as the newly discovered hurdle in output connectivity, the lack of myelination. Candidate as well as unbiased approaches are pursued to identify the critical checkpoints and overcome them.