Summary
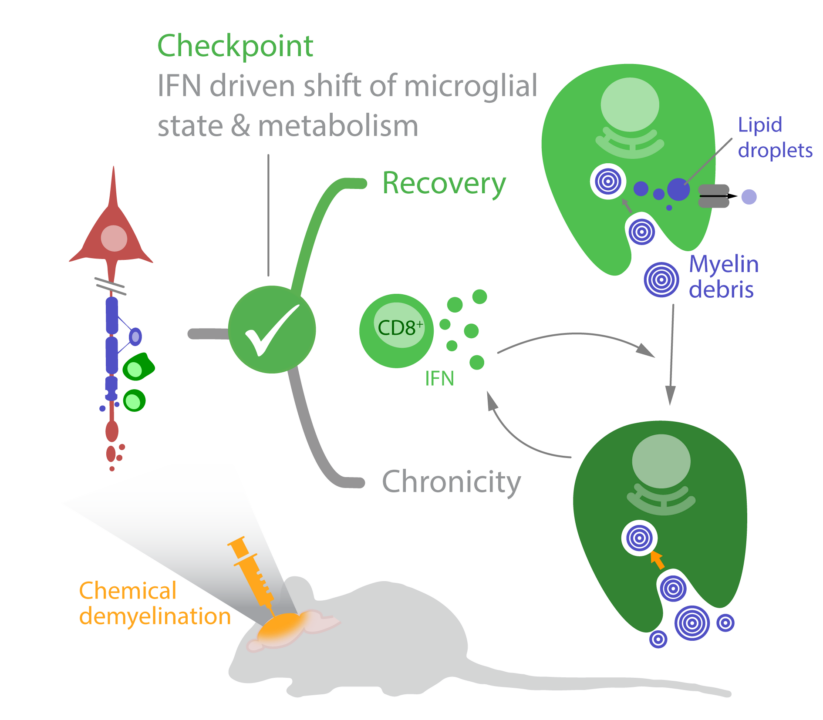
Demyelinating injuries in the central nervous system trigger a regenerative process that relies on coordinated responses from multiple cell types, including the innate immune system. Here, we propose that a maladaptive chronic immune response may be initiated when microglia’s capacity to clear myelin debris is overwhelmed, leading to a self-propagating immune response that impairs remyelination and causes further tissue damage. We aim to identify the molecular checkpoints in microglia/macrophages that drive this process, with a focus on interferon signaling.