Summary
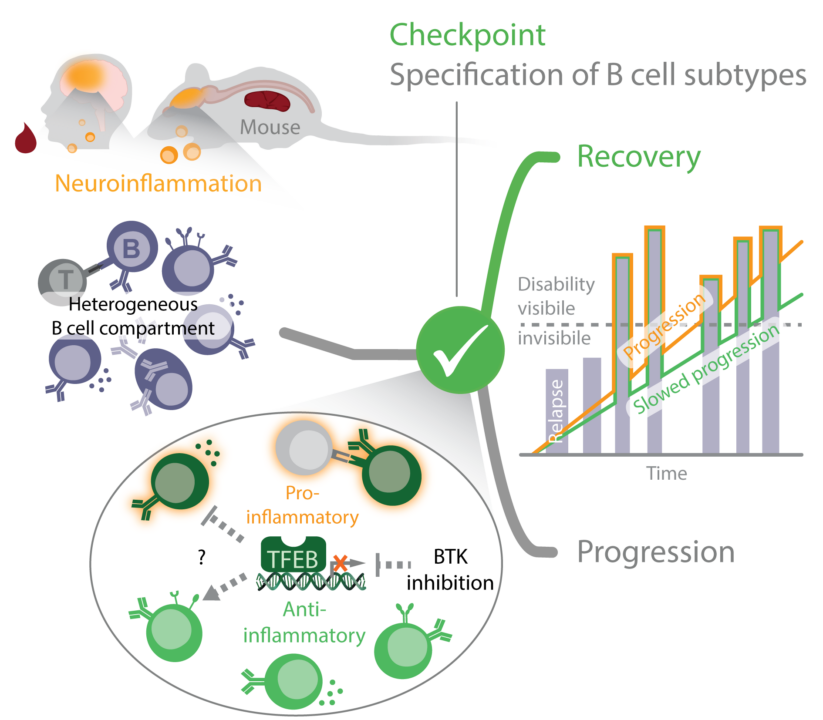
B lymphocytes have been more recently recognized to possess a dual role in MS pathogenesis. On the one hand, B cells promote inflammation of the central nervous system and MS progression, while on the other hand, B-lymphoid inhibition of pathogenic T cell functions ameliorates MS severity and facilitates recovery from acute MS bouts. To understand the functional dichotomy, we aim at a comprehensive phenotyping of individual B cell subpopulations followed by their transcriptional profiling to better understand the molecular checkpoints of B cell specification.




